Mastering Medical Device Compliance: Your End-to-End FDA/ISO Audit Management Workflow
Publicado: Actualizado: 04/16/2026
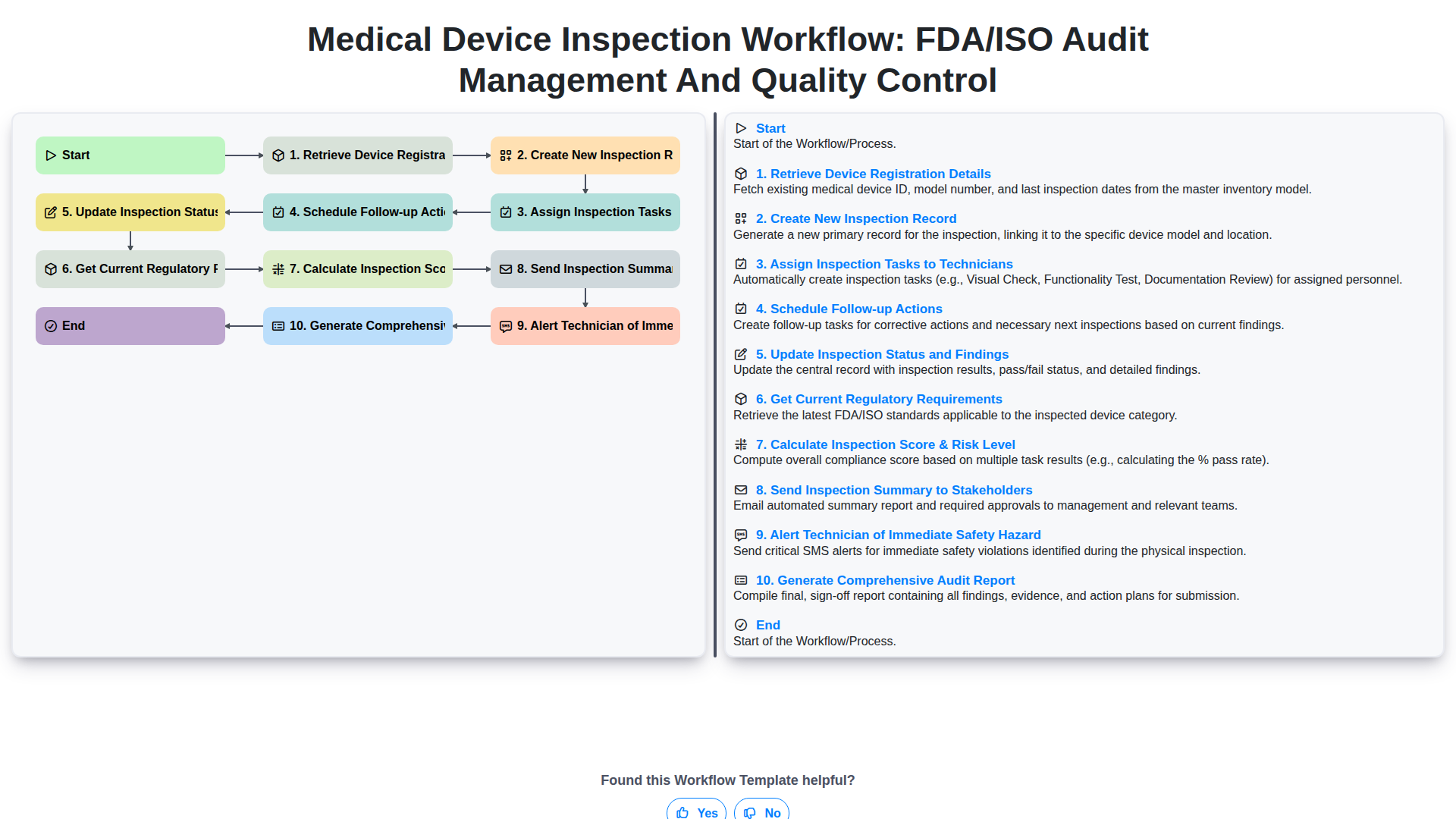
Índice
- Introduction: The Criticality of Streamlined Audit Management
- Step 1: Establishing the Foundation - Retrieving Device Registration Details
- Step 2: Initiating the Audit Lifecycle - Creating the New Inspection Record
- Step 3: Execution Efficiency - Assigning Inspection Tasks to Technicians
- Step 4: Continuous Improvement - Scheduling Follow-up Actions
- Step 5: Real-Time Visibility - Updating Inspection Status and Findings
- Step 6: Staying Ahead of Change - Integrating Current Regulatory Requirements (FDA/ISO)
- Step 7: Risk Assessment in Practice - Calculating the Inspection Score & Risk Level
- Step 8: Prioritizing Safety - Implementing Immediate Hazard Alerts for Technicians
- Step 9: Communication Hub - Sending Inspection Summary to Key Stakeholders
- Step 10: Closure and Documentation - Generating the Comprehensive Audit Report
- Best Practices for Maintaining Workflow Integrity
- The Technology Backbone: Choosing the Right Audit Management System
- Conclusion: From Compliance Burden to Operational Advantage
- Resources & Links
TLDR: This guide walks you through a structured, end-to-end workflow template designed to streamline and de-risk FDA/ISO compliance audits for medical devices. By following these steps-from initial data retrieval and task assignment to final reporting and risk calculation-you ensure every inspection is managed systematically, keeping your quality control robust, efficient, and audit-ready.
Introduction: The Criticality of Streamlined Audit Management
In the highly regulated landscape of medical device manufacturing, compliance is not merely a suggestion-it is a fundamental necessity for patient safety and market access. Navigating the demands of bodies like the FDA and adhering to international standards such as ISO requires meticulous documentation, rigorous process control, and flawless execution during inspections. A manual, disparate, or ad-hoc approach to audit management invariably leads to bottlenecks, potential compliance gaps, and significant delays. Effective audit management is thus more than just ticking boxes; it is a proactive system designed to maintain an uninterrupted state of quality control. A standardized, streamlined workflow ensures that every step, from initial preparation to final report generation, is tracked, verified, and executed against the latest regulatory mandates.
Step 1: Establishing the Foundation - Retrieving Device Registration Details
The journey into managing a medical device inspection begins with meticulous preparation, and this starts with comprehensive knowledge of the product itself. The very first critical step in our workflow is the retrieval of the Device Registration Details. This isn't just about pulling up a serial number; it's about building a complete historical and technical profile of the device under inspection. By systematically gathering information such as the device's unique Model Number, Unique Device Identification (UDI), initial submission date, current regulatory classification (e.g., Class II, Class III), and the specific predicate devices, we establish our baseline. This data forms the immutable foundation upon which every subsequent audit decision, task assignment, and compliance check will be built. Incorrect or incomplete registration data at this stage can lead to flawed risk assessments and costly delays, making this verification point non-negotiable for maintaining audit integrity.
Step 2: Initiating the Audit Lifecycle - Creating the New Inspection Record
This stage marks the official commencement of the inspection process. Once the initial trigger for an audit-whether it's a routine surveillance check, a response to a complaint, or an investigation into a specific finding-is identified, the system must facilitate the creation of a dedicated, comprehensive Inspection Record. This record is the central digital repository for the entire audit, housing everything from the scope of work to the final corrective action plan. Key details to be captured here include the unique device ID, the specific regulatory standards being assessed (e.g., 21 CFR Part 820, ISO 13485), and the initial date and anticipated duration of the inspection. Accurate initial data entry at this point prevents scope creep and ensures that all subsequent activities are tied to a traceable, verifiable audit instance, forming the foundational backbone for all subsequent quality management activities.
Step 3: Execution Efficiency - Assigning Inspection Tasks to Technicians
Once the inspection record is created, the crucial next step is ensuring the right expertise reaches the right place. This involves systematically assigning specific inspection tasks to qualified technicians. This isn't just about handing out a checklist; it requires mapping the complexity of the device and the specific areas under review to the unique skill sets, certifications, and availability of your technical team. Intelligent workflow management systems allow you to automate this assignment process, ensuring that specialized tasks-like electrical safety testing or biocompatibility reviews-are immediately routed to technicians with proven proficiency in those domains. Furthermore, the system should track technician workloads to prevent bottlenecks, guaranteeing that no critical inspection task stalls due to over-assignment or underutilization of personnel.
Step 4: Continuous Improvement - Scheduling Follow-up Actions
This crucial step transforms a mere inspection finding into tangible quality improvements. Simply recording a deficiency is insufficient; the system must proactively manage the remediation lifecycle. Scheduling follow-up actions involves logging specific corrective and preventive actions (CAPA) required for each identified deviation. This includes assigning owners, setting hard deadlines, and defining necessary supporting documentation. Automated reminders, integrated with the technician's task list, ensure that no critical follow-up falls through the cracks. By treating the follow-up as a distinct, tracked workflow, organizations ensure accountability and measure the efficacy of their corrective measures, driving the entire quality system toward perpetual excellence.
Step 5: Real-Time Visibility - Updating Inspection Status and Findings
This stage is where the entire process gains its critical heartbeat: real-time, actionable visibility. As inspections progress, continuous updates to the inspection record are paramount. Technicians, upon completing a task or identifying a deviation, must immediately update the inspection status within the system. This shouldn't be a batch update at the end; it must be granular. For instance, if a sample test is inconclusive, the finding-including supporting evidence (photos, logs, preliminary analysis)-is uploaded immediately. The system must flag this finding, linking it directly to the relevant section of the device registration and the specific regulatory standard it impacts. This instant documentation ensures that the audit trail is immutable and that management has an up-to-the-minute view of both the progress made and the issues discovered.
Step 6: Staying Ahead of Change - Integrating Current Regulatory Requirements (FDA/ISO)
A critical aspect of maintaining compliance is ensuring your inspection process doesn't become obsolete. The regulatory landscape is constantly shifting, with the FDA and ISO standards regularly updating their guidelines. Therefore, the workflow must incorporate a dedicated step to Get Current Regulatory Requirements. This isn't a mere checklist addition; it requires active integration. Your system should not only prompt the user to check the latest guidelines but ideally, it should pull or alert based on known changes. For instance, if a significant revision occurs in ISO 13485 concerning risk management, the system should flag every active inspection record and prompt the assigned Quality Manager to review the impact assessment against the previous standard. This proactive step minimizes the risk of finding out after the audit that a procedure was technically non-compliant with the newest iteration of the standard.
Step 7: Risk Assessment in Practice - Calculating the Inspection Score & Risk Level
This step moves the workflow from pure data collection to active risk management. The calculated Inspection Score and resulting Risk Level are critical decision points that dictate the immediate next steps, the urgency of corrective actions, and the overall compliance posture of the device or process under review. The scoring mechanism isn't just arithmetic; it's a weighted assessment that synthesizes findings from previous steps-such as deviations noted during the technical inspection, adherence to historical quality benchmarks, and the severity of any safety hazards flagged. A low score signals a need for comprehensive remediation plans, whereas a high score might warrant an immediate hold on the product line pending deeper investigation. This data point becomes the core input for executive decision-making, ensuring that limited QA resources are always directed to the areas presenting the greatest potential patient risk or regulatory exposure.
Step 8: Prioritizing Safety - Implementing Immediate Hazard Alerts for Technicians
This step is a critical deviation from standard procedural documentation and moves into active, real-time risk management. When an inspection uncovers an immediate threat to patient safety-such as identifying a critical failure in sterilization protocols or an unmitigated design flaw-the process must bypass standard reporting queues. The workflow must be engineered to trigger an instantaneous, high-priority alert directly to the assigned technician, regardless of what other tasks are being processed (e.g., documentation updates or scheduling). This alert is not just a notification; it is an actionable stop signal. The alert mechanism must be multi-layered, utilizing SMS, in-app pop-ups, and potentially even audible warnings within the inspection station interface. Upon triggering, the system must simultaneously log the alert timestamp, the specific hazard identified, and the technician who received it, ensuring an auditable trail proving that the warning was disseminated instantly. This proactive safety alert capability is a hallmark of a mature, robust quality system, demonstrating a commitment that outweighs mere compliance checking.
Step 9: Communication Hub - Sending Inspection Summary to Key Stakeholders
This crucial step ensures that all relevant parties are immediately informed about the outcomes of the inspection. The system must facilitate the seamless distribution of the compiled inspection summary-which includes findings, identified deviations, and the calculated risk level-to stakeholders such as Quality Assurance Managers, Executive Leadership, R&D Heads, and Regulatory Affairs personnel. Automated, role-based distribution is key here. The summary shouldn't just be an attachment; it should be an interactive digest within the communication channel, allowing stakeholders to immediately click on specific findings, view associated evidence, and acknowledge receipt, creating an undeniable audit trail of communication and understanding.
Step 10: Closure and Documentation - Generating the Comprehensive Audit Report
This final, crucial step solidifies the entire inspection process by transforming collected data into actionable intelligence. Generating the comprehensive audit report is more than just compiling documents; it's synthesizing findings into a narrative that demonstrates compliance, identifies systemic risks, and provides a clear roadmap for improvement. This report must serve as the definitive record of the audit. It typically includes a summary of the initial scope, detailed findings categorized by severity (e.g., Critical, Major, Minor), evidence supporting each finding (including photos, logs, and procedural deviations), and the outcomes of all corrective and preventive actions (CAPAs) discussed. For regulatory bodies like the FDA or ISO assessors, this report provides the auditable trail that proves due diligence. It's essential that the report clearly distinguishes between observations, non-conformances, and positive findings, ensuring all stakeholders-management, engineering, quality, and leadership-understand the overall health and compliance posture of the device and the associated processes. A well-structured report doesn't just state what happened; it explains why it happened and how the organization plans to prevent it from happening again.
Best Practices for Maintaining Workflow Integrity
Consistent adherence to established protocols is the cornerstone of robust workflow integrity. Instead of treating each inspection as a standalone event, organizations must view the entire process as a continuous feedback loop. This means that the findings documented during one inspection-such as a recurring procedural gap or a necessary equipment calibration-must automatically trigger preventative measures or updates to the baseline documentation for the next inspection. Furthermore, centralizing documentation and making it immediately accessible to all authorized personnel prevents version control issues, ensuring that technicians are always working from the most current iteration of SOPs and regulatory guidelines. Regular, cross-functional training that simulates potential workflow breaks is also paramount, transforming potential points of failure into muscle memory for the team.
The Technology Backbone: Choosing the Right Audit Management System
Selecting the right Audit Management System (AMS) is not merely an IT purchase; it's a foundational investment in your compliance infrastructure. In the highly regulated world of medical devices, the AMS must be more than a digital checklist-it needs to be an intelligent workflow engine. A robust AMS should seamlessly manage the entire lifecycle of an inspection, from initial registration to final reporting. Ideally, the system will allow you to programmatically Retrieve Device Registration Details to ensure every inspection starts with an accurate, up-to-date baseline. It must facilitate the creation of granular, traceable records, allowing you to Create New Inspection Records and immediately Assign Inspection Tasks to Technicians, ensuring accountability from minute one. Furthermore, the best systems don't just document actions; they anticipate them, enabling automated triggers to Schedule Follow-up Actions and providing a centralized dashboard to Update Inspection Status and Findings in real-time. Crucially, these platforms integrate real-time data feeds, allowing them to ingest and cross-reference the Current Regulatory Requirements from global sources, which is vital for context-aware inspection. Advanced AMS solutions go further by providing analytical power, capable of analyzing collected data to Calculate Inspection Score & Risk Level instantly, moving you from reactive compliance to proactive risk management. Finally, the system must streamline communication, enabling immediate Alert Technician of Immediate Safety Hazard alerts, generating a final Comprehensive Audit Report, and automating the distribution of the Inspection Summary to Stakeholders-all within a controlled, auditable digital pathway.
Conclusion: From Compliance Burden to Operational Advantage
Ultimately, viewing medical device inspection and regulatory management solely as a compliance burden is a narrow perspective. By implementing a structured, digitized workflow-like the one encompassing device registration, task assignment, continuous status updates, risk scoring, and comprehensive reporting-organizations fundamentally shift their operational paradigm. This systematic approach transforms necessary adherence into a measurable operational advantage. It doesn't just check compliance boxes; it proactively builds quality into the process, accelerates corrective actions, mitigates unforeseen risks before they become critical failures, and solidifies a culture of unwavering quality assurance.
Resources & Links
- U.S. Food and Drug Administration (FDA) : The primary source for all US medical device regulations, guidance documents, and compliance standards.
- International Organization for Standardization (ISO) : The global body for developing and publishing international standards, including those relevant to Quality Management Systems (e.g., ISO 13485).
- Medical Device Industry News/Resources : General industry news and best practice articles regarding quality system implementation and audits.
- Regulatory Compliance Consulting Firms : Resources from firms specializing in FDA/ISO compliance, offering practical workflow management insights.
- Healthcare IT & Compliance Software Review Sites : Articles reviewing Computerized Quality Management Systems (QMS) and audit management software.
- Biomedical Science/Quality Assurance Associations : Educational resources on quality assurance best practices for life sciences and medical devices.
Preguntas frecuentes
What is the main goal of an end-to-end audit management workflow?
The primary goal is to transition from a reactive approach to a proactive one, ensuring continuous readiness for both FDA inspections and ISO audits by managing documentation, risk assessment, and corrective actions within a unified system.
What is the difference between an FDA inspection and an ISO audit in medical device compliance?
An FDA inspection is a regulatory enforcement action focused on verifying compliance with 21 CFR 820 (Quality System Regulation) and ensuring public safety, whereas an ISO audit (such as ISO 13485) is a third-party certification process focused on adherence to international quality management system standards.
What are the key stages of a successful audit management workflow?
A robust workflow includes pre-audit preparation (gap analysis and internal audits), the audit execution phase (managing findings and observations), and the post-audit phase (CAPA implementation, root cause analysis, and documentation closure).
How does CAPA (Corrective and Preventive Action) integrate into the audit workflow?
CAPA is the critical closing loop in the workflow; once an audit identifies a non-conformance, the CAPA process must be triggered to investigate the root cause, implement fixes, and verify that the solution prevents recurrence.
What role does documentation play in maintaining audit readiness?
Documentation is the foundation of compliance; maintaining accurate, traceable, and up-to-date records (such as Design History Files and Device Master Records) is essential to prove that all regulatory and quality processes were followed as intended.
¿Te resultó útil este artículo?
Demostración de la solución de gestión de auditorías/inspecciones
¡Asegure el cumplimiento y mejore el rendimiento! ChecklistGuro simplifica la creación, ejecución e informes de auditorías/inspecciones. Reduzca riesgos, mejore la calidad y mantenga la consistencia. Administre todo con nuestro Work OS.
Artículos relacionados
Restaurant Kitchen Safety Inspection Checklist Template
The Ultimate Brewery Equipment Sanitation & Safety Checklist Template
Plantilla de verificación para la inspección de aerogeneradores: Su guía completa
The Ultimate Apartment Building Maintenance Inspection Checklist Template
Your Essential Guide to a Manufacturing Quality Control Inspection Checklist
Plantilla Definitiva de Lista de Verificación para Inspección de Paneles Solares
Your Ultimate HVAC Inspection Checklist Template
La plantilla definitiva de lista de verificación para inspecciones de centros de datos.
Podemos hacerlo juntos
¿Necesita ayuda con las listas de verificación?
¿Tienes alguna pregunta? Estamos aquí para ayudarte. Envía tu consulta y te responderemos a la brevedad.