Streamlining Success: The Ultimate Medical Device Inspection Workflow for Auditing and Quality Management
Publicado: Actualizado: 04/16/2026
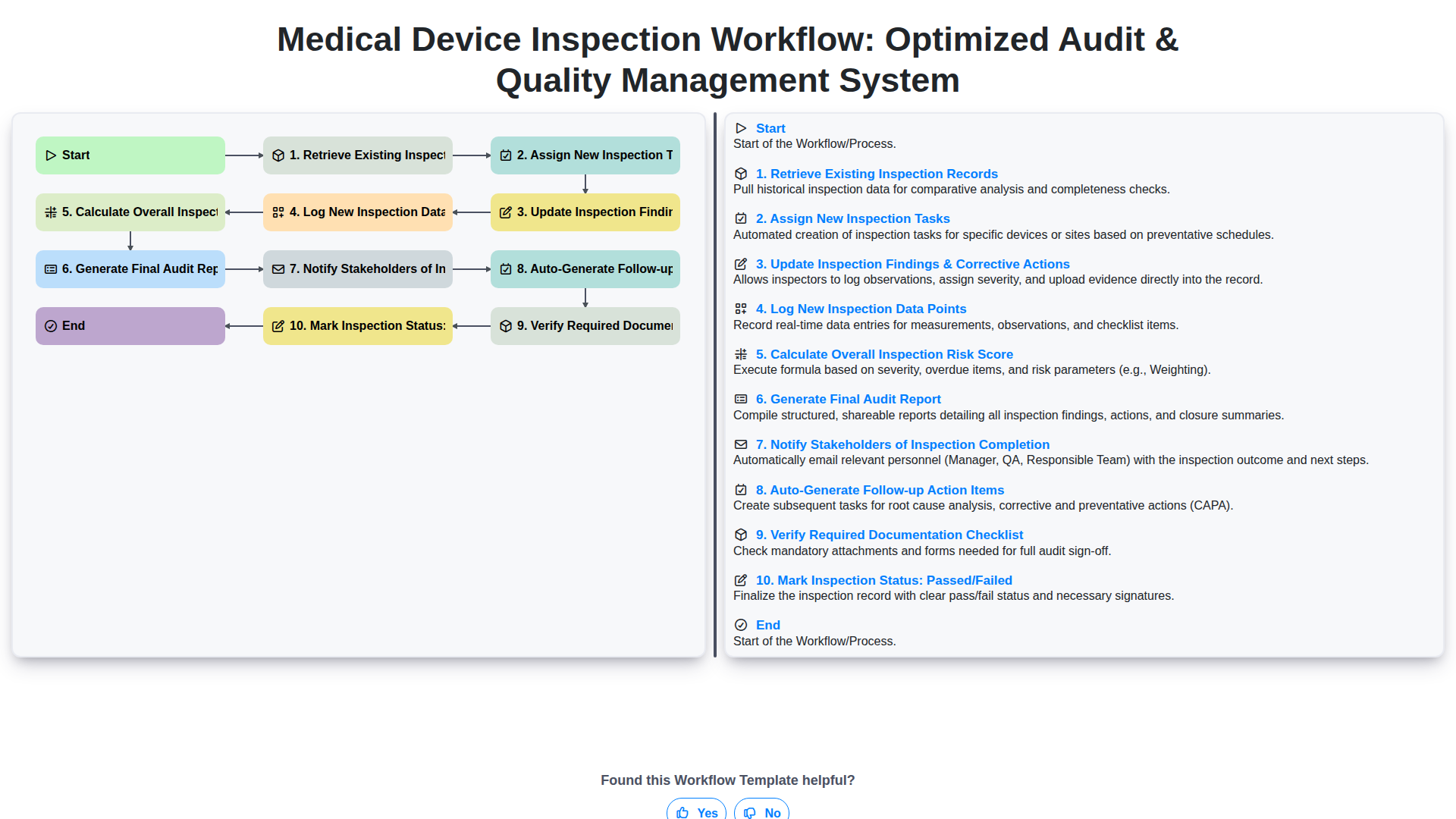
Índice
- Introduction: The Imperative of a Robust Inspection Workflow
- Step 1: Foundation - Retrieving Existing Inspection Records for Context
- Step 2: Proactive Auditing - Efficiently Assigning New Inspection Tasks
- Step 3: Continuous Improvement - Updating Inspection Findings & Corrective Actions (CAPA)
- Step 4: Data Capture Excellence - Logging New Inspection Data Points Accurately
- Step 5: Predictive Risk Management - Calculating the Overall Inspection Risk Score
- Step 6: Synthesis - Generating the Comprehensive Final Audit Report
- Step 7: Closing the Loop - Notifying Stakeholders of Inspection Completion
- Step 8: Ensuring Compliance - Auto-Generating Follow-up Action Items
- Step 9: Due Diligence - Verifying Required Documentation Checklist
- Step 10: Final Decision Point - Marking Inspection Status: Passed/Failed
- Workflow Optimization: Selecting the Right Technology Stack
- Benefits of a Streamlined Medical Device Inspection Workflow
- Best Practices for Maintaining Workflow Integrity and Audit Readiness
- Conclusion: Driving Quality and Safety Through Optimized Processes
- Resources & Links
TLDR: This workflow provides a comprehensive, step-by-step system to manage medical device inspections, from retrieving historical records and assigning new tasks to calculating risk scores, documenting findings, and automatically generating reports and follow-up actions, ensuring compliance and consistent quality management.
Introduction: The Imperative of a Robust Inspection Workflow
In the highly regulated and safety-critical world of medical device manufacturing, the integrity of the quality management system (QMS) is not just a requirement-it's a matter of patient safety. Inspections, whether internal audits, supplier assessments, or regulatory site visits, are the backbone of verifying compliance. However, the traditional, manual processes surrounding these inspections are notoriously cumbersome, prone to human error, and slow to adapt. Reworking audit and quality management is no longer an optional improvement; it's an operational necessity. A disjointed workflow leads to lost data, missed follow-up actions, and an inflated risk profile for the device and the company.
Step 1: Foundation - Retrieving Existing Inspection Records for Context
The journey of any comprehensive quality management system begins with a deep understanding of what has come before. Before tackling a new audit or inspection, the first critical step is to retrieve and thoroughly review existing inspection records. This isn't merely an archival task; it's the foundation upon which optimized inspection protocols are built. By accessing historical data, your team can immediately establish a baseline of compliance, identify recurring process weaknesses, and understand the historical performance of specific equipment, departments, or procedures. This context is invaluable, allowing inspectors to move beyond superficial checks and focus their attention on high-risk areas flagged in past reports. Reviewing past Corrective and Preventive Actions (CAPAs) also reveals established trends-showing whether past interventions have been fully effective or if systemic issues persist, ensuring that the current inspection is proactive rather than purely reactive.
Step 2: Proactive Auditing - Efficiently Assigning New Inspection Tasks
This stage is where the process shifts from reviewing the past to actively shaping the future of quality assurance. Instead of waiting for compliance deadlines, the system allows teams to proactively anticipate potential failure points. By assigning new inspection tasks digitally, you eliminate manual assignment errors and delays. The workflow engine can utilize historical data, identified high-risk components, or scheduled maintenance cycles to automatically populate the task queue. This ensures that inspections are performed not just when they are due, but when they are most needed, maximizing uptime and minimizing the risk of unplanned compliance gaps.
Step 3: Continuous Improvement - Updating Inspection Findings & Corrective Actions (CAPA)
This stage is where the true depth of quality management shines. It moves the process beyond mere compliance to genuine improvement. Once initial findings are documented, the focus shifts entirely to the Corrective and Preventive Action (CAPA) loop. For every identified deviation, finding, or non-conformance, a robust action must be initiated. This isn't just about logging a problem; it's about root cause analysis (RCA). Our system facilitates this by guiding the user through structured RCA methodologies-be it the 5 Whys or Fishbone Diagrams-ensuring the team doesn't just treat the symptom. The Updating Inspection Findings & Corrective Actions module acts as the central hub where the initial finding is cross-referenced against established protocols, the RCA is documented, the corrective action plan (CAP) is drafted, assigned owners, and due dates are set. Crucially, the system tracks the implementation status of these actions, preventing them from falling into shelfware. This iterative updating ensures that the gap found during the audit is systematically closed, monitored for effectiveness, and documented, transforming a single inspection into a powerful driver for organizational quality maturity.
Step 4: Data Capture Excellence - Logging New Inspection Data Points Accurately
This stage is the heart of real-time quality control. Accurately logging every piece of data encountered during the physical inspection is non-negotiable for compliance and continuous improvement. Our optimized workflow guides inspectors through structured data entry points, ensuring nothing is missed. This includes documenting readings from calibrated measurement tools, recording batch traceability numbers, noting environmental conditions (temperature, humidity), and capturing photographic or video evidence directly into the system. By standardizing data capture-whether it's a pass/fail metric, a specific deviation observed, or a machine serial number-we move beyond subjective notes and create an objective, auditable data trail. Integrating smart forms and mandatory field requirements minimizes human error at the source, ensuring that the data collected is immediately useful for later analysis and risk calculation.
Step 5: Predictive Risk Management - Calculating the Overall Inspection Risk Score
This step moves beyond simple pass/fail reporting to provide proactive, quantitative insight into the device's quality standing. Instead of just cataloging past issues, the system calculates a comprehensive, Overall Inspection Risk Score. This score is not merely an average; it's a weighted, predictive index derived from inputs across the entire workflow-including the severity and recurrence of past findings, the number of outstanding Corrective Actions (CAPAs), the adherence rate to documentation checks, and the complexity of the associated device model. A high risk score doesn't just signal danger; it immediately directs quality management attention to the most vulnerable processes, allowing teams to intervene before a critical failure occurs. This predictive capability shifts the quality function from being purely reactive (fixing what broke) to being inherently proactive (preventing what might break).
Step 6: Synthesis - Generating the Comprehensive Final Audit Report
This crucial phase transforms raw data and disparate actions into a single, cohesive narrative of the inspection's outcome. The system doesn't just compile data; it synthesizes it. It pulls together the initial findings from updated records, integrates the logged data points, and contextualizes the calculated overall risk score. The resulting Final Audit Report is far more than a collection of scores; it's a management tool. It clearly articulates what was found, why it represents a risk based on predefined metrics, and what needs to be done next. This comprehensive report serves as the official historical record, providing executive summaries, detailed evidence trails, and management sign-off sections, ensuring that all stakeholders receive a unified, actionable document rather than scattered reports.
Step 7: Closing the Loop - Notifying Stakeholders of Inspection Completion
Once the audit report is finalized and the immediate corrective actions are documented, the final critical step is formal communication. This isn't just an email; it's a structured notification process ensuring every relevant party is immediately aware of the outcome. The system automatically triggers targeted alerts to department heads, Quality Assurance officers, and relevant executive management. This notification includes a high-level summary of the findings, the final pass/fail status, and clearly links to the comprehensive audit report. This immediate closure ensures transparency, allows stakeholders to begin resource allocation for any required follow-up training or systemic changes right away, and officially transitions the task record to Closed, maintaining an accurate audit trail of accountability.
Step 8: Ensuring Compliance - Auto-Generating Follow-up Action Items
This critical step transforms a completed inspection from a mere report into a pathway for continuous improvement. Instead of relying on manual follow-up, the system automatically analyzes the findings, non-conformances, and risk scores generated throughout the audit. If a critical finding is recorded, the system immediately triggers relevant, pre-defined corrective and preventive action (CAPA) items. This proactive approach ensures that no identified vulnerability is forgotten. The action items are automatically logged into the CAPA module, assigned to the responsible department or individual, and given a preliminary due date based on the severity of the finding. This immediate workflow prevents backlogs, keeps compliance momentum high, and guarantees accountability right from the moment the last clipboard is signed off.
Step 9: Due Diligence - Verifying Required Documentation Checklist
This crucial stage ensures that no gaps exist in your audit trail or compliance records. Before finalizing the inspection, the system must prompt the user to verify against a comprehensive, pre-defined documentation checklist. This isn't merely a suggestion; it's a mandatory gate check. The workflow should flag any missing documents-such as updated biocompatibility reports, latest sterilization validation logs, or specific training certifications-and prevent progression to the final reporting stage until these gaps are addressed, providing immediate, actionable feedback to the inspector.
Step 10: Final Decision Point - Marking Inspection Status: Passed/Failed
This is the critical juncture where all the preceding data, analysis, and actions converge into a definitive outcome. The 'Mark Inspection Status: Passed/Failed' step formalizes the conclusion of the entire audit process. Based on the aggregated findings, the severity of non-conformities recorded, the completeness of the corrective actions, and the risk score calculated, the system must make a clear, justifiable determination. If all critical checkpoints are cleared and identified risks are mitigated according to pre-defined thresholds, the status is marked 'Passed.' Conversely, if significant deviations, unresolved critical findings, or inadequate responses are noted, the status is marked 'Failed.' This status update isn't merely a tag; it triggers subsequent workflow escalations-a 'Failed' status automatically initiates urgent remediation tracks, whereas 'Passed' confirms compliance and allows the process to move toward final closure and certification.
Workflow Optimization: Selecting the Right Technology Stack
To truly optimize a medical device inspection process, the technology stack must be as robust and interconnected as the workflow it aims to manage. Relying on disparate spreadsheets, emails, and physical binders is not merely inefficient; it's a compliance risk. The ideal stack needs to move beyond mere data collection and facilitate genuine process intelligence. This means adopting a centralized, cloud-based platform-preferably one built specifically for Quality Management Systems (QMS) or regulated industries. Such a system must offer integrated modules that handle everything from initial task assignment to final report generation, ensuring data integrity at every handoff point. Furthermore, the stack must support advanced functionality like AI-driven risk scoring and automated deviation tracking, transforming the system from a digital filing cabinet into a proactive risk mitigation tool.
Benefits of a Streamlined Medical Device Inspection Workflow
A streamlined workflow moves beyond simply documenting inspections; it actively improves compliance, reduces risk, and saves valuable time. By automating sequential steps-from retrieving past records to generating final reports-organizations minimize the risk of human error, ensuring that every critical data point is captured accurately. Furthermore, integrating risk scoring and automated follow-up ensures that corrective actions aren't just recommended, but are actioned promptly, leading to a more robust Quality Management System (QMS) overall.
Best Practices for Maintaining Workflow Integrity and Audit Readiness
Maintaining workflow integrity is not just about following a process; it's about ensuring the accuracy and completeness of the data trail that underpins your quality management system. For medical device inspection workflows, where compliance gaps can have significant ramifications, strict adherence to best practices is crucial for audit readiness. First and foremost, establish clear ownership for each step. When multiple teams touch an inspection record-from initial task assignment to final report generation-the process must define who is responsible for verification at each handoff point. Secondly, leverage digital workflows to minimize manual data entry, which is a leading cause of human error. Automation should handle repetitive tasks like populating timestamps, version control, and basic data logging. Furthermore, implementing robust version control for both the SOPs (Standard Operating Procedures) and the inspection records themselves is non-negotiable. This ensures that any finding recorded against a device or process is linked to the exact version of the guideline that was active at the time of the inspection. Finally, conduct regular dry-run audits of your workflow, simulating a real audit scenario without an actual finding. This proactive testing helps you identify bottlenecks, gaps in documentation, or ambiguous decision points before a regulator or client finds them for you.
Conclusion: Driving Quality and Safety Through Optimized Processes
By systematically implementing an optimized medical device inspection workflow, organizations move beyond mere compliance and build a proactive culture of quality and safety. This streamlined process-from retrieving historical data and efficiently assigning new tasks to calculating comprehensive risk scores and generating actionable follow-up items-ensures that every audit contributes tangible value. Ultimately, adopting such a robust, digitized system minimizes human error, accelerates decision-making, and provides an unparalleled level of transparency. This isn't just about passing an audit; it's about embedding continuous improvement into the core DNA of your quality management system, ensuring the highest standard of patient safety is maintained across your entire product lifecycle.
Resources & Links
- U.S. Food and Drug Administration (FDA) Guidelines : Official regulatory guidance and guidelines for medical device quality system requirements, essential for workflow context.
- ISO/IEC Standards Body (e.g., ISO 13485) : Key international standards for medical device quality management systems that dictate workflow necessity.
- Medical Device QMS Software Vendors : Websites showcasing modern Quality Management System (QMS) platforms capable of automating inspection workflows.
- FDA Quality System Regulation (21 CFR Part 820) : The foundational regulations governing the design, manufacture, and servicing of medical devices.
- Market Research Reports on Audit Technology : Reports detailing the technological trends and best practices in digital auditing and compliance management.
- FDA Inspection and Audit Process Information : Understanding the typical scope and nature of regulatory inspections to model a realistic workflow.
- Industry Peer Journals/Publications : Professional journals offering case studies and deep dives into process optimization within MedTech.
Preguntas frecuentes
What is the primary goal of a streamlined medical device inspection workflow?
The primary goal is to enhance efficiency and accuracy in identifying non-conformities, ensuring regulatory compliance, and maintaining high standards of product safety and quality management.
Who are the key stakeholders involved in the medical device inspection process?
Key stakeholders include Quality Assurance (QA) teams, regulatory affairs specialists, production managers, internal and external auditors, and regulatory bodies such as the FDA or EMA.
How does an optimized inspection workflow impact regulatory compliance?
An optimized workflow ensures that all inspection steps are standardized, documented, and traceable, which reduces the risk of human error and ensures readiness for audits by demonstrating adherence to ISO 13485 and other regulatory requirements.
What are the essential components of an effective inspection workflow?
Essential components include clearly defined inspection criteria, standardized testing protocols, real-time data capture, automated reporting, and a robust corrective and preventive action (CAPA) system.
How can technology assist in streamlining the auditing process?
Technology, such as automated inspection software and digital checklists, can streamline auditing by enabling real-time data monitoring, reducing manual documentation errors, and providing instant access to historical compliance data.
What is the role of CAPA in the medical device inspection workflow?
Corrective and Preventive Action (CAPA) is critical for addressing the root causes of any non-conformities identified during inspection, preventing their recurrence, and driving continuous improvement in the manufacturing process.
¿Te resultó útil este artículo?
Demostración de la solución de gestión de auditorías/inspecciones
¡Asegure el cumplimiento y mejore el rendimiento! ChecklistGuro simplifica la creación, ejecución e informes de auditorías/inspecciones. Reduzca riesgos, mejore la calidad y mantenga la consistencia. Administre todo con nuestro Work OS.
Artículos relacionados
Restaurant Kitchen Safety Inspection Checklist Template
The Ultimate Brewery Equipment Sanitation & Safety Checklist Template
Plantilla de verificación para la inspección de aerogeneradores: Su guía completa
The Ultimate Apartment Building Maintenance Inspection Checklist Template
Your Essential Guide to a Manufacturing Quality Control Inspection Checklist
Plantilla Definitiva de Lista de Verificación para Inspección de Paneles Solares
Your Ultimate HVAC Inspection Checklist Template
La plantilla definitiva de lista de verificación para inspecciones de centros de datos.
Podemos hacerlo juntos
¿Necesita ayuda con las listas de verificación?
¿Tienes alguna pregunta? Estamos aquí para ayudarte. Envía tu consulta y te responderemos a la brevedad.