Computer System Validation (CSV) Workflow
Streamline your regulatory compliance with our optimized Computer System Validation (CSV) Workflow. Designed specifically for the pharmaceutical industry, this end-to-end process ensures seamless GxP compliance, rigorous data integrity, and audit-ready documentation throughout the entire system lifecycle-from User Requirements (URS) to final Summary Reports. Eliminate manual errors and accelerate technology deployment while meeting stringent FDA 21 CFR Part 11 and EU Annex 11 standards.
Start
Start of the Workflow/Process.
1. Fetch System Requirements
Retrieve all User Requirements (URS) entries from the Requirements Data Model to initiate the validation scope.
2. Create Validation Plan Task
Assign a task to the QA Lead to draft the Validation Plan based on the identified system scope.
3. Generate Validation Plan Entry
Create a new entry in the Validation Plan data model containing the scope, strategy, and testing approach.
4. Risk Assessment Task
Assign a task to the Subject Matter Expert (SME) to perform a GAMP5-based risk assessment.
5. Create Risk Assessment Record
Create an entry in the Risk Management data model to document identified hazards and mitigation strategies.
6. Calculate Risk Priority Number (RPN)
Execute formula (Severity * Occurrence * Detectability) to determine the critical level of each risk.
7. Update Risk Mitigation Status
Update the Risk Assessment entry to reflect that mitigation controls have been implemented.
8. Execute IQ/OQ/PQ Testing
Assign testing tasks to Validation Engineers to execute Installation, Operational, and Performance Qualifications.
9. Create Test Script Entry
Create a record in the Test Script data model for each protocol being executed.
10. Log Test Results
Update the Test Script entry with pass/fail results and evidence links after execution.
11. Summarize Deviation Count
Aggregate the number of 'Fail' statuses from all Test Script entries to determine total deviations.
12. Investigate Deviations Task
Create a task for the Quality Engineer if the Deviation Count is greater than zero.
13. Create Deviation Report
Create an entry in the Deviation Data Model for every failed test case identified.
14. Close Deviations
Update the Deviation entries to 'Closed' once corrective actions are verified.
15. Generate Traceability Matrix Report
Create a report linking Requirements $\rightarrow$ Risks $\rightarrow$ Test Scripts to ensure full coverage.
16. Final Summary Report Review
Assign a task to the Quality Manager to review the final Validation Summary Report (VSR).
17. Create Validation Summary Report
Create the final record in the Validation Summary data model summarizing the entire process outcome.
18. Notify Stakeholders of Approval
Send an email to the Project Sponsor and IT Department once the VSR is approved and the system is released for production.
19. Update System Status to 'Validated'
Update the Computer System entry in the Inventory Data Model to 'Validated' status.
End
End of the Workflow/Process.
Start of the Workflow/Process.
Retrieve all User Requirements (URS) entries from the Requirements Data Model to initiate the validation scope.
Assign a task to the QA Lead to draft the Validation Plan based on the identified system scope.
Create a new entry in the Validation Plan data model containing the scope, strategy, and testing approach.
Assign a task to the Subject Matter Expert (SME) to perform a GAMP5-based risk assessment.
Create an entry in the Risk Management data model to document identified hazards and mitigation strategies.
Execute formula (Severity * Occurrence * Detectability) to determine the critical level of each risk.
Update the Risk Assessment entry to reflect that mitigation controls have been implemented.
Assign testing tasks to Validation Engineers to execute Installation, Operational, and Performance Qualifications.
Create a record in the Test Script data model for each protocol being executed.
Update the Test Script entry with pass/fail results and evidence links after execution.
Aggregate the number of 'Fail' statuses from all Test Script entries to determine total deviations.
Create a task for the Quality Engineer if the Deviation Count is greater than zero.
Create an entry in the Deviation Data Model for every failed test case identified.
Update the Deviation entries to 'Closed' once corrective actions are verified.
Create a report linking Requirements $\rightarrow$ Risks $\rightarrow$ Test Scripts to ensure full coverage.
Assign a task to the Quality Manager to review the final Validation Summary Report (VSR).
Create the final record in the Validation Summary data model summarizing the entire process outcome.
Send an email to the Project Sponsor and IT Department once the VSR is approved and the system is released for production.
Update the Computer System entry in the Inventory Data Model to 'Validated' status.
End of the Workflow/Process.
Found this Workflow Template helpful?
Pharmaceutical Management Solution Demo
Navigating complex regulations and ensuring quality in pharmaceutical manufacturing? ChecklistGuro's Work OS platform streamlines processes from R&D to production, packaging, and distribution. Maintain compliance, improve efficiency, and reduce risk. Discover how ChecklistGuro can transform your pharmaceutical operations!
Related Workflow Templates

Regulatory Submission Dossier Management

Environmental Monitoring and Cleanroom Control
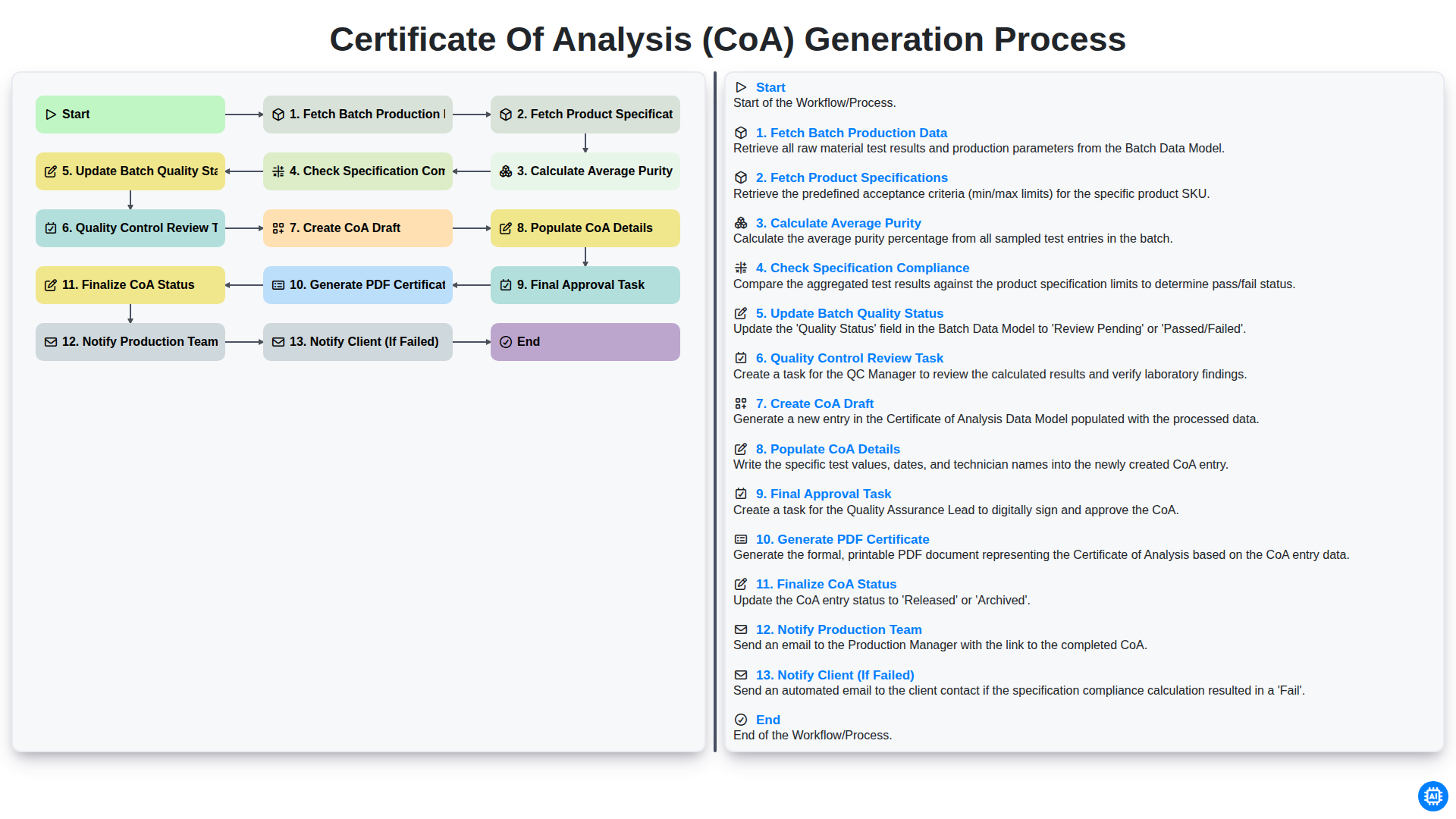
Certificate of Analysis (CoA) Generation Process

Equipment Calibration and Maintenance Management
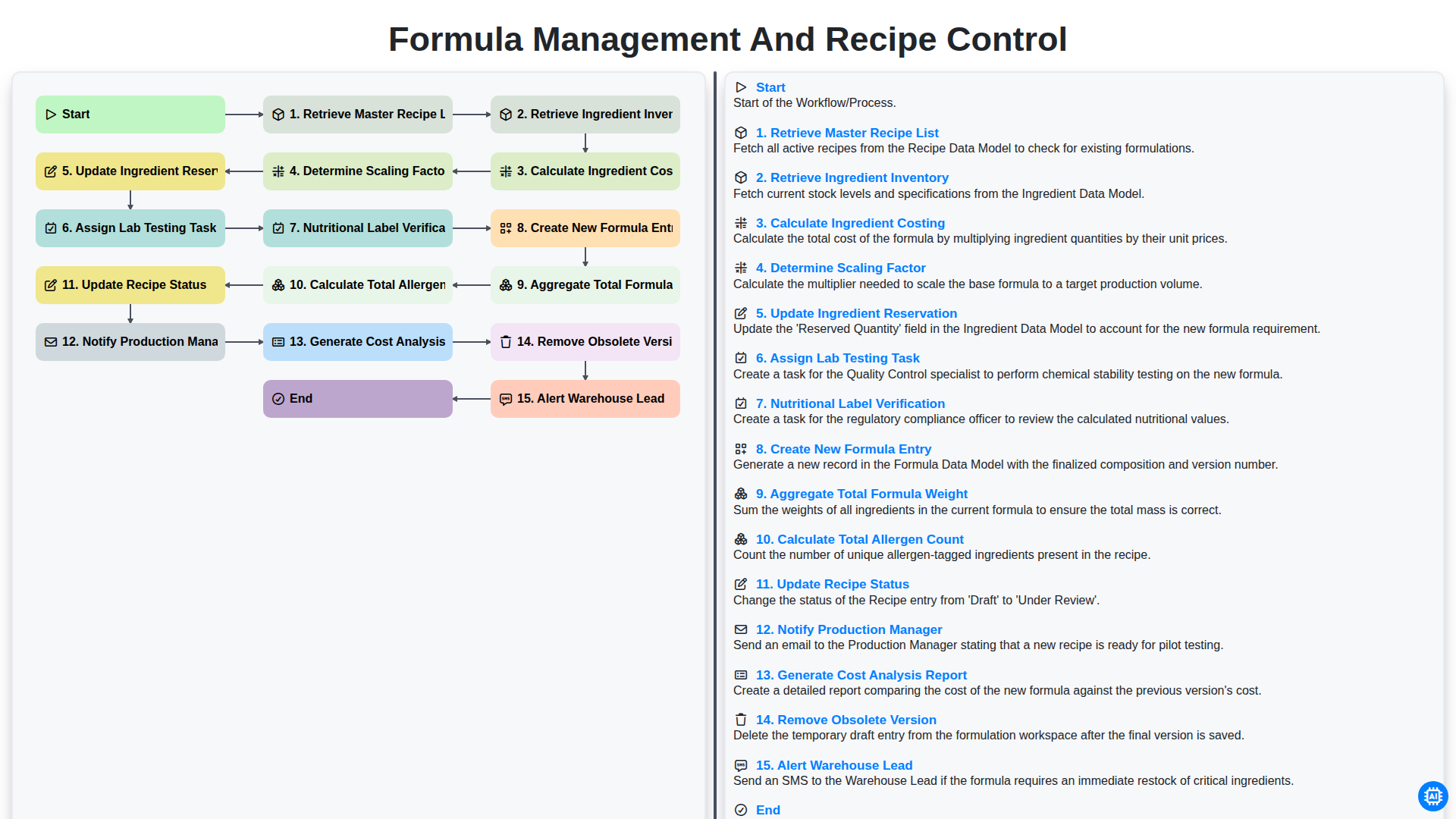
Formula Management and Recipe Control

Regulatory Labeling and Packaging Control
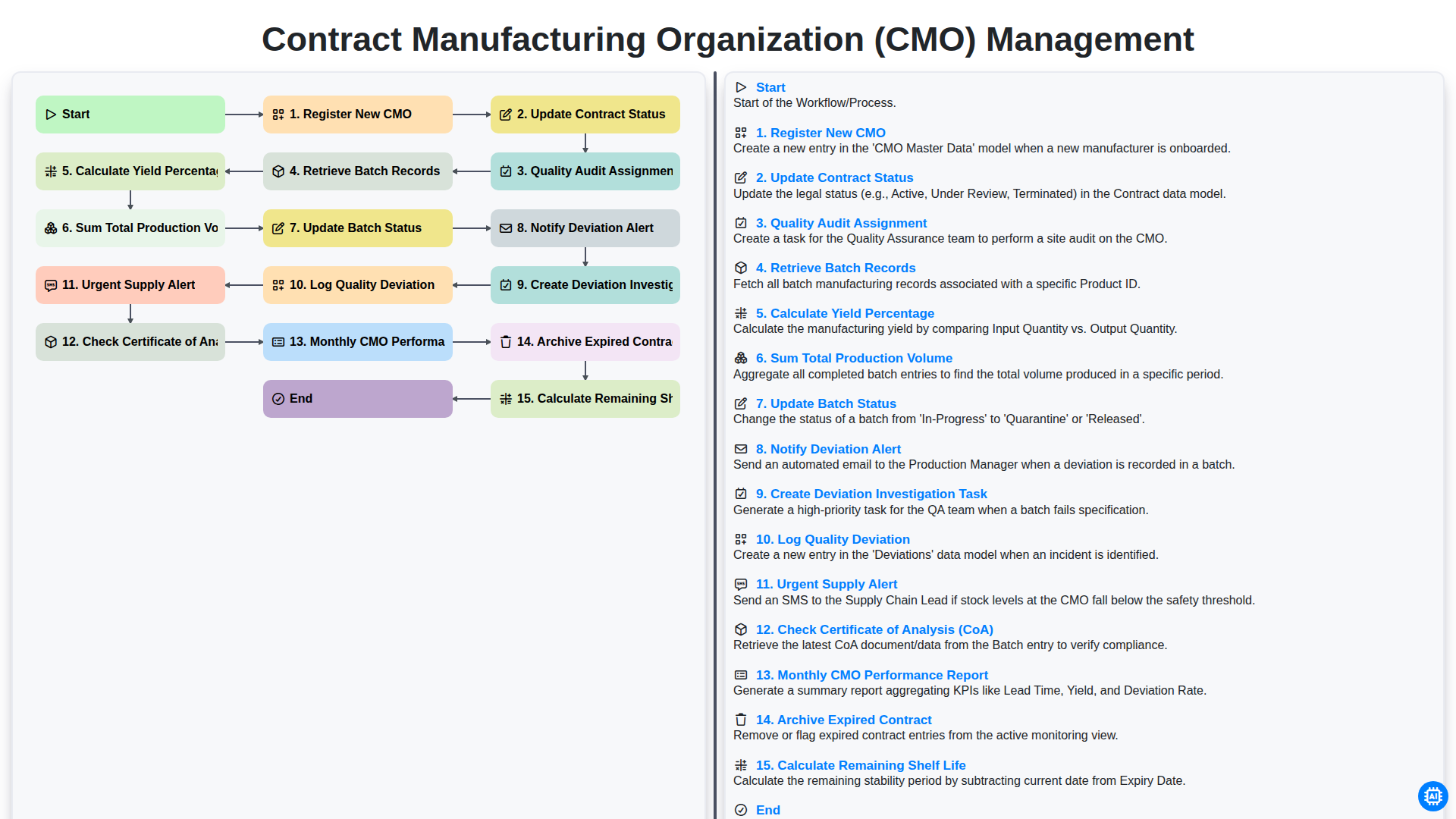
Contract Manufacturing Organization (CMO) Management
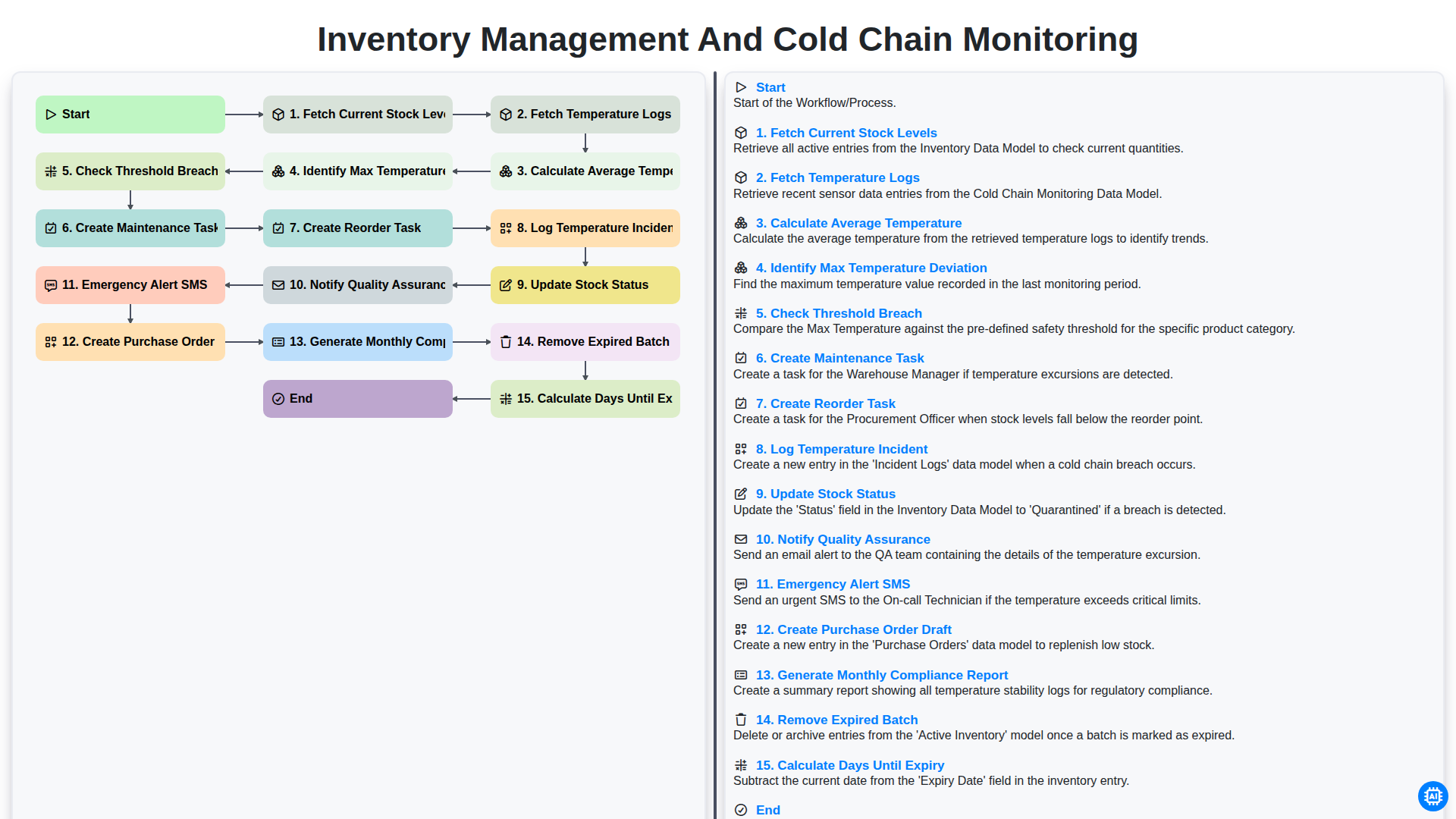
Inventory Management and Cold Chain Monitoring
We can do it Together
Need help with
Pharmaceutical?
Have a question? We're here to help. Please submit your inquiry, and we'll respond promptly.