Medical Device Inspection Workflow: Best Practices for FDA/ISO Audit Management
Published: 06/04/2026 Updated: 06/05/2026
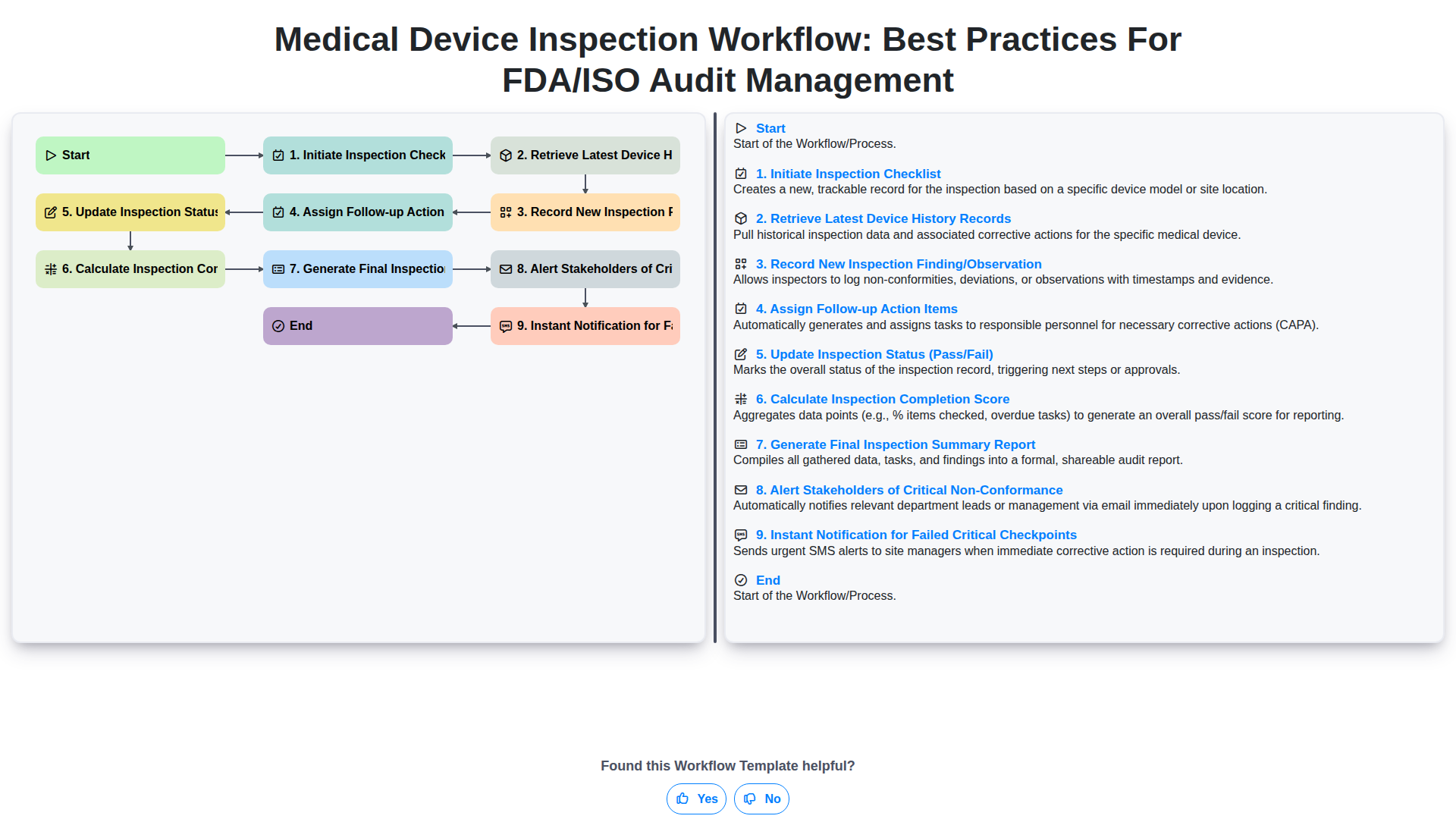
Table of Contents
- Introduction: The Critical Role of Standardized Inspection Workflows
- The Foundation of Compliance: Initiating the Inspection Checklist
- Ensuring Data Integrity: Retrieving the Latest Device History Records (DHR)
- Real-Time Documentation: Recording New Inspection Findings and Observations
- Driving Accountability: Assigning Follow-up Action Items
- Defining Outcomes: Updating Inspection Status (Pass/Fail)
- Quantifying Quality: Calculating the Inspection Completion Score
- Closing the Loop: Generating the Final Inspection Summary Report
- Proactive Risk Mitigation: Alerting Stakeholders of Critical Non-Conformance
- Automating Urgency: Instant Notifications for Failed Critical Checkpoints
- Best Practices for Maintaining FDA and ISO Audit Readiness
- Conclusion: Building a Continuous Improvement Culture
- Resources & Links
TLDR: Streamline your regulatory compliance and audit readiness with a structured medical device inspection workflow. This guide explores a systematic approach-from initiating checklists and reviewing Device History Records to managing non-conformances and automated stakeholder alerts-ensuring your inspection process meets rigorous FDA and ISO standards while minimizing human error.
Introduction: The Critical Role of Standardized Inspection Workflows
In the highly regulated landscape of medical device manufacturing, the margin for error is zero. When navigating the complexities of FDA 21 CFR Part 820 and ISO 13485 standards, compliance is not merely a goal-it is a prerequisite for market access and patient safety. At the heart of this compliance lies the inspection process.
A fragmented or manual inspection process is more than just an operational inefficiency; it is a significant regulatory risk. Inconsistent documentation, missed observations, and delayed follow-up actions can lead to much larger systemic failures, resulting in costly warning letters, product recalls, or even complete shutdowns during an audit.
A standardized, end-to-end inspection workflow serves as the backbone of a robust Quality Management System (QMS). By transforming inspections from a series of disconnected tasks into a continuous, automated, and traceable loop, manufacturers can ensure that every device meets predefined specifications and that every non-conformance is addressed with surgical precision. Implementing a structured workflow doesn't just prepare you for an audit-it makes audit readiness a natural byproduct of your daily operations.
The Foundation of Compliance: Initiating the Inspection Checklist
The integrity of a medical device inspection begins long before a physical examination takes place. The first, and arguably most critical, step in a robust compliance workflow is the Initiation of the Inspection Checklist. This phase serves as the blueprint for the entire audit process, ensuring that every regulatory requirement-whether mandated by FDA 21 CFR Part 820 or ISO 13485-is accounted for.
An effective initiation process is not merely about opening a document; it is about establishing a standardized, repeatable framework that eliminates human error. By initiating a formal checklist, quality assurance teams can define the scope of the inspection, identify the specific device batches or production lines under review, and ensure that the criteria for compliance are clearly documented from the outset.
A well-structured initiation phase sets the stage for the subsequent retrieval of Device History Records (DHRs) and ensures that the inspectors are working from a single source of truth. Without this foundational step, inspections become reactive and disorganized, significantly increasing the risk of missed non-conformances and costly regulatory findings during official audits.
Ensuring Data Integrity: Retrieving the Latest Device History Records (DHR)
At the heart of any successful medical device inspection lies the integrity of your documentation. The step of retrieving the latest Device History Records (DHR) is perhaps the most critical moment in the workflow, as it serves as the single source of truth for the inspection process. In an era of strict FDA and ISO compliance, relying on outdated, fragmented, or unverified paper logs is a high-risk maneuver that can lead to catastrophic audit findings.
To ensure regulatory readiness, the inspection workflow must be integrated with a real-time data retrieval system. This ensures that inspectors are evaluating the most recent version of the manufacturing, processing, and testing data, including all recent modifications, quality control sign-offs, and material certifications.
When the workflow automatically pulls the most current DHR, it eliminates the version control trap-where an inspector inadvertently validates a device against obsolete specifications. This seamless retrieval process guarantees that the data being analyzed is synchronized with the current state of production, providing the foundational accuracy required to defend your quality management system during a formal audit. In short, you cannot inspect what you cannot accurately verify; retrieving the latest DHR is the safeguard that ensures your inspection is based on reality, not outdated snapshots.
Real-Time Documentation: Recording New Inspection Findings and Observations
In the high-stakes environment of medical device manufacturing, the gap between identifying a non-conformance and documenting it can be the difference between a seamless audit and a major regulatory finding. Real-time documentation is not merely a matter of convenience; it is a fundamental pillar of data integrity and compliance with FDA 21 CFR Part 11 and ISO 13485 standards.
The Record New Inspection Finding/Observation step in an optimized workflow moves away from the outdated practice of manual, paper-based logging. Instead, it leverages digital interfaces that allow inspectors to capture deviations, anomalies, or out-of-specification (OOS) results at the exact moment they are discovered. By digitizing this process, organizations eliminate the risks associated with illegible handwriting, lost notes, and retrospective logging-a practice that frequently triggers red flags during regulatory inspections.
Effective real-time recording should capture more than just a description of the error. A robust digital workflow enables inspectors to attach photographic evidence, link specific measurement values, and timestamp each entry automatically. This creates an immutable audit trail that provides a granular view of the inspection event. When every observation is recorded instantly, the Single Source of Truth is maintained, ensuring that the inspection data is accurate, contemporaneous, and ready for immediate scrutiny by quality assurance teams and regulatory auditors alike.
Driving Accountability: Assigning Follow-up Action Items
An inspection is only as valuable as the actions taken in its aftermath. Identifying a deviation is a critical milestone, but the true test of a robust Quality Management System (QMS) lies in how a facility responds to that discovery. Without a structured process for Assigning Follow-up Action Items, findings often languish in spreadsheets, leading to recurring non-conformances and increased regulatory risk.
To drive true accountability, the transition from finding to fixing must be seamless and highly visible. A best-practice workflow ensures that once an observation is recorded, the system automatically triggers the creation of actionable tasks. These tasks should be explicitly linked to the specific non-conformance and assigned to a designated owner with a clear, mandatory deadline.
By integrating follow-up assignments directly into your inspection workflow, you move away from passive documentation and toward proactive remediation. This approach ensures that CAPAs (Corrective and Preventive Actions) are initiated immediately, preventing minor discrepancies from escalating into major audit findings during FDA or ISO inspections. In short, accountability is no longer a manual chase-it is a built-in feature of your inspection lifecycle.
Defining Outcomes: Updating Inspection Status (Pass/Fail)
At the conclusion of every inspection cycle, the workflow must culminate in a definitive determination: Pass or Fail. This step is more than just a simple checkbox; it is the formal closure of the inspection event and the foundation upon which all subsequent regulatory compliance decisions are built.
An accurate status update serves as the single source of truth for your quality management system. A Pass status signifies that the device meets all predefined criteria and is cleared for the next stage of the lifecycle, such as release or distribution. Conversely, a Fail status triggers an immediate halt, signaling that the device or batch does not meet the necessary safety or efficacy standards and requires immediate intervention.
To maintain audit readiness, this update must be:
- Data-Driven: The status should be a direct reflection of the Calculate Inspection Completion Score step, ensuring that the decision is based on objective metrics rather than subjective intuition.
- Immutable and Traceable: In the eyes of FDA and ISO auditors, a status change must be accompanied by a clear digital audit trail, documenting exactly when the decision was made and by which authorized personnel.
- Action-Oriented: The transition to a Fail status should act as the automated catalyst for the next phases of the workflow, such as initiating non-conformance reports (NCRs) and notifying stakeholders.
By standardizing how you define and record these outcomes, you eliminate ambiguity, reduce the risk of human error, and ensure that your facility maintains a high standard of regulatory integrity.
Quantifying Quality: Calculating the Inspection Completion Score
In a high-stakes regulatory environment, subjective assessments of good or bad are insufficient. To move beyond intuition and toward true data-driven quality management, organizations must implement a standardized method for calculating the inspection completion score.
This metric serves as a real-time quantitative indicator of device compliance and manufacturing health. Rather than viewing an inspection as a simple binary outcome, a completion score aggregates the performance of individual checkpoints into a single, measurable value. This is typically achieved by assigning weighted values to each item on your inspection checklist-where critical-to-quality (CTQ) parameters carry higher weightings than minor cosmetic checks.
By quantifying inspection results, quality managers can:
- Identify Trends Over Time: Track whether your completion score is improving or degrading across different production lots or manufacturing shifts.
- Benchmark Performance: Compare inspection outputs across multiple production lines to identify specific areas requiring systemic corrective actions.
- Enable Predictive Maintenance: A downward trend in completion scores can serve as an early warning signal for equipment fatigue or process drift before a formal non-conformance occurs.
Ultimately, the inspection completion score transforms raw inspection data into actionable intelligence, allowing your team to move from a reactive find and fix mindset to a proactive predict and prevent strategy.
Closing the Loop: Generating the Final Inspection Summary Report
The culmination of a successful inspection process is the generation of the Final Inspection Summary Report. This document serves as the single source of truth for both internal quality management and external regulatory bodies. Rather than manually compiling data from disparate spreadsheets or paper logs, an automated workflow ensures that this report is a real-time, accurate reflection of the inspection's outcome.
A robust reporting engine automatically aggregates all data points collected throughout the workflow-from the initial checklist execution to the final calculated completion score. By pulling directly from the Device History Records (DHR) and the recorded inspection findings, the system eliminates human error and the risk of data transcription discrepancies.
The final report should provide a high-level overview for management while offering deep-dive granularity for engineers. It must clearly outline:
- Compliance Status: A definitive Pass/Fail verdict based on predefined critical checkpoints.
- Quantitative Metrics: The final inspection completion score, providing a benchmark for quality trends.
- Deficiency Summaries: A detailed log of all observations and the specific follow-up actions assigned to rectify them.
By automating this stage, your team moves from data collection to data utilization. The summary report doesn't just close the current inspection loop; it provides the foundational data needed for trend analysis, CAPA (Corrective and Preventive Actions) triggers, and seamless preparation for the next FDA or ISO audit.
Proactive Risk Mitigation: Alerting Stakeholders of Critical Non-Conformance
In the high-stakes environment of medical device manufacturing, timing is the most critical factor in preventing a minor deviation from escalating into a catastrophic regulatory failure. A proactive risk mitigation strategy relies on moving away from reactive, manual notifications toward an automated, real-time alerting system.
When a critical non-conformance is detected during the inspection process, the window for effective intervention is incredibly small. Waiting for a manual end-of-shift review can mean the difference between a simple corrective action and a costly product recall or a formal FDA Warning Letter. By implementing automated triggers, your workflow can instantly bridge the gap between detection and response.
Effective risk mitigation involves two layers of automated communication:
- Stakeholder Alerts for Critical Non-Conformance: As soon as a non-conformance is identified that meets specific critical criteria, the system immediately notifies key stakeholders-including Quality Assurance Managers, Production Leads, and Regulatory Affairs officers. This ensures that the personnel responsible for high-level decision-making are informed the moment a risk to product safety or compliance is identified.
- Instant Notifications for Failed Critical Checkpoints: For high-risk parameters-such as sterilization integrity, biocompatibility markers, or structural dimensions-the workflow should trigger an immediate, instantaneous alert. These stop-gap notifications act as a digital circuit breaker, preventing the device from moving further down the production line until the failure is addressed.
By automating these alerts, you eliminate human error and communication delays. This ensures that your team is never surprised by a quality issue during an audit, but rather, is already actively managing the resolution before the auditor even steps onto the floor.
Automating Urgency: Instant Notifications for Failed Critical Checkpoints
In the high-stakes environment of medical device manufacturing, time is the most critical variable. When a critical checkpoint fails, the window for corrective action is incredibly narrow. Manual notification processes-such as sending emails, making phone calls, or relying on physical logbooks-introduce dangerous latency that can lead to uncontained non-conformances and significant regulatory risk.
Automating your workflow transforms delayed awareness into immediate intervention. By integrating Instant Notifications for Failed Critical Checkpoints, your system acts as a 24/7 digital sentry. The moment an inspector marks a critical parameter as Fail, the system triggers an automated alert to the Quality Assurance (QA) manager, production leads, and regulatory officers.
This real-time trigger ensures that:
- Containment happens instantly: Production lines can be halted or quarantined before defective units move further down the supply chain.
- The Detection-to-Correction gap is minimized: Engineers can begin root cause analysis while the inspection data is still fresh.
- Audit Trails are synchronized: The notification itself becomes a timestamped part of the digital record, demonstrating to FDA and ISO auditors that your organization maintains proactive, rather than reactive, oversight.
By eliminating the human element from the notification loop, you ensure that critical errors are never buried in an inbox or missed during a shift change, maintaining the integrity of your quality management system.
Best Practices for Maintaining FDA and ISO Audit Readiness
Maintaining continuous audit readiness is no longer a seasonal task; it is a fundamental requirement for staying compliant with FDA 21 CFR Part 820 and ISO 13485 standards. To transition from a reactive firefighting mode to a proactive state of compliance, organizations must move away from fragmented spreadsheets and toward a standardized, integrated inspection workflow.
The following best practices are essential for ensuring your inspection processes stand up to the scrutiny of rigorous regulatory audits:
- Standardize the Inspection Trigger: Audit readiness begins before the inspection starts. By implementing a formal process to Initiate Inspection Checklists, you ensure that every inspector follows a uniform, validated protocol, reducing the risk of human error or overlooked requirements.
- Ensure Data Integrity via Single Source of Truth: An inspection is only as reliable as the data behind it. Always Retrieve the Latest Device History Records (DHR) as the foundational step. Using the most current, version-controlled documentation prevents findings based on obsolete information, which is a frequent red flag for auditors.
- Real-Time Documentation of Deviations: One of the most critical aspects of ISO compliance is the immediacy of recording. You must Record New Inspection Findings or Observations at the moment they occur. Delayed documentation leads to memory bias and loss of critical context, potentially undermining the validity of your entire quality system.
- Close the Loop with Actionable Accountability: Finding a non-conformance is only half the battle; the regulatory focus is on how you remediate it. Implement a system to Assign Follow-up Action Items with clear ownership and deadlines. This transforms an observation into a closed-loop CAPA (Corrective and Preventive Action) process.
- Quantifiable Compliance Monitoring: To maintain a high-level view of quality health, you should Update Inspection Status (Pass/Fail) and Calculate Inspection Completion Scores automatically. These metrics provide the objective evidence required during audits to prove that your quality oversight is consistent and measurable.
- Automated Escalation and Communication: In a regulated environment, silence is a risk. High-stakes failures require immediate visibility. Establishing Instant Notifications for Failed Critical Checkpoints and Alerting Stakeholders of Critical Non-Conformance ensures that your engineering and management teams can intervene before a minor deviation escalates into a major non-compliance or a product recall.
- Standardized Reporting for Audit Evidence: At the conclusion of every cycle, Generate a Final Inspection Summary Report. A structured, automated report serves as a permanent, unalterable record that demonstrates to FDA investigators that your inspection process is controlled, documented, and followed to completion.
Conclusion: Building a Continuous Improvement Culture
Mastering a standardized medical device inspection workflow is more than just a strategy for regulatory compliance; it is a fundamental driver of operational excellence. By transitioning from reactive, manual processes to an automated, data-driven inspection ecosystem, manufacturers move beyond merely surviving audits to actively thriving through quality intelligence.
The true value of implementing a structured workflow-from initiating checklists to real-time stakeholder alerts-lies in the ability to transform raw inspection data into actionable insights. When every non-conformance is instantly recorded, every follow-up action is tracked, and every failure triggers immediate notification, the organization develops a closed-loop quality system. This visibility allows leadership to identify recurring trends, address root causes before they escalate into systemic issues, and foster an environment where quality is everyone's responsibility.
Ultimately, an efficient inspection workflow serves as the backbone of a continuous improvement culture. It empowers your team to maintain the highest standards of patient safety and product efficacy, ensuring that your organization remains audit-ready every single day, rather than just once a year.
Resources & Links
- FDA - Medical Devices: The official website of the U.S. Food and Drug Administration, providing regulatory guidelines, CFR Title 21 regulations, and compliance standards for medical device manufacturers.
- ISO - Medical Devices: The International Organization for Standardization, providing access to ISO 13485 standards and global quality management system requirements for the medical device industry.
- American Society for Quality (ASQ): A global resource for quality professionals offering best practices in inspection, auditing, and quality control methodologies essential for manufacturing excellence.
- Regulatory Affairs Professionals Society (RAPS): A professional community providing resources, news, and training on navigating the complex regulatory landscapes of global medical device markets.
- International Society for Pharmaceutical Engineering (ISPE): Provides technical resources and best practices regarding much of the manufacturing processes, including device history records and quality management integration.
- Institute of Internal Auditors (IIA): Resources for establishing robust internal audit workflows, managing non-conformance reporting, and maintaining institutional accountability.
Found this Article helpful?
Audit/Inspection Management Solution Demo
Ensure compliance & improve performance! ChecklistGuro streamlines audit/inspection creation, execution, and reporting. Reduce risk, enhance quality, & maintain consistency. Manage it all with our Work OS.
Related Articles
Restaurant Kitchen Safety Inspection Checklist Template
The Ultimate Brewery Equipment Sanitation & Safety Checklist Template
Wind Turbine Inspection Checklist Template: Your Comprehensive Guide
The Ultimate Apartment Building Maintenance Inspection Checklist Template
Your Essential Guide to a Manufacturing Quality Control Inspection Checklist
The Ultimate Solar Panel Inspection Checklist Template
Your Ultimate HVAC Inspection Checklist Template
The Ultimate Data Center Inspection Checklist Template
We can do it Together
Need help with
Checklists?
Have a question? We're here to help. Please submit your inquiry, and we'll respond promptly.