Pharmaceutical GMP Inspection Workflow: Validated Compliance and Audit Management
Published: 06/04/2026 Updated: 06/05/2026
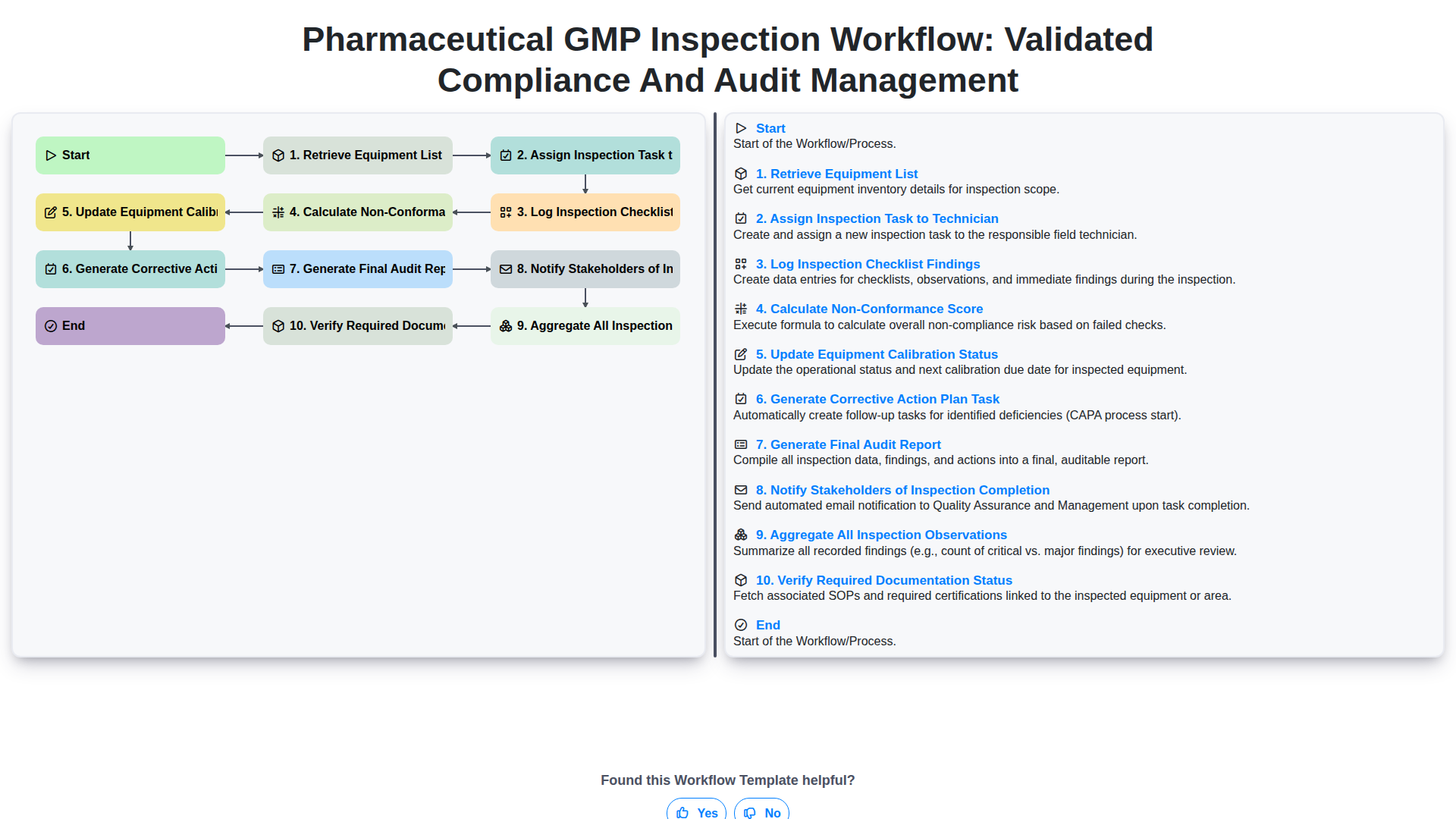
Table of Contents
- Introduction to GMP Inspection Workflows
- The Critical Role of Validated Compliance in Pharmaceutical Manufacturing
- Phase 1: Preparatory Steps and Resource Allocation
- Initiating the Process: Retrieving Equipment Lists
- Task Delegation: Assigning Inspection Roles to Technicians
- Phase 2: On-Site Inspection and Data Capture
- Real-Time Documentation: Logging Inspection Checklist Findings
- Phase 3: Risk Assessment and Quantitative Analysis
- Quantifying Deviations: Calculating Non-Conformance Scores
- Phase 4: Post-Inspection Remediation and Updates
- Maintaining Equipment Integrity: Updating Calibration Status
- Closing the Loop: Generating Corrective Action Plans (CAPA)
- Phase 5: Reporting, Verification, and Stakeholder Communication
- Conclusion: Ensuring Audit Readiness through Systematic Management
- Resources & Links
TLDR: Streamline your regulatory readiness with this automated Pharmaceutical GMP Inspection Workflow. This guide explains how to transition from manual tracking to a validated, end-to-end process-covering everything from technician task assignment and real-time checklist logging to automated non-conformance scoring and final audit report generation-ensuring continuous compliance and efficient audit management.
Introduction to GMP Inspection Workflows
In the highly regulated landscape of pharmaceutical manufacturing, maintaining Good Manufacturing Practice (GMP) is not merely a regulatory obligation but the foundation of patient safety and product integrity. At the heart of this-compliance lies the inspection workflow-a structured, systematic approach to verifying that equipment, processes, and environments meet stringent quality standards.
A robust GMP inspection workflow serves as a continuous loop of oversight, ensuring that every piece of critical machinery and every operational process is scrutinized against validated protocols. Rather than being a sporadic event, an effective workflow integrates real-time data collection, rigorous error scoring, and immediate corrective action integration. By transforming inspection from a manual, fragmented task into a unified digital process, manufacturers can move away from reactive troubleshooting and toward a state of audit-ready excellence. This structured approach ensures that every observation is captured, every non-conformance is quantified, and every stakeholder is informed, ultimately safeguarding the lifecycle of the product and the trust of the end consumer.
The Critical Role of Validated Compliance in Pharmaceutical Manufacturing
In the highly regulated landscape of pharmaceutical manufacturing, the margin for error is non-existent. Compliance is not merely a regulatory requirement; it is the foundation of patient safety and product efficacy. Within this high-stakes environment, validated compliance serves as the structural integrity of the entire production lifecycle. It ensures that every process, from raw material intake to final packaging, is executed according to predefined, rigorous standards that are documented, reproducible, and verifiable.
The challenge lies in the fact that compliance is not a static achievement but a continuous state of vigilance. This is where audit management becomes indispensable. A robust, validated workflow prevents the fragmentation of data and the dangerous silos of information that often lead to regulatory citations. By integrating real-time monitoring with standardized inspection protocols, manufacturers can move from a reactive stance-responding to deviations after they occur-to a proactive posture of inspection readiness.
Effective audit management ensures that every observation is tracked, every non-conformance is quantified, and every corrective action is closed with traceable evidence. In an era of increasing scrutiny from global regulatory bodies like the FDA and EMA, leveraging a structured, validated workflow is the only way to ensure that compliance remains an inherent feature of the manufacturing process rather than a secondary, manual effort.
Phase 1: Preparatory Steps and Resource Allocation
The foundation of a successful pharmaceutical GMP inspection lies in meticulous preparation. Before any physical inspection takes place, the process must begin with Retrieving the Equipment List, which serves as the definitive roadmap for the audit. This step ensures that every piece of machinery, from autoclaves to tablet presses, is accounted for and that no critical asset is overlooked during the assessment.
Once the scope is defined, the workflow moves into the critical stage of Assigning Inspection Tasks to Technicians. In a GMP environment, competency is non-negotiable; therefore, delegating specific inspection zones or equipment types to qualified personnel ensures that the audit is conducted with the necessary technical expertise. This phase also includes the essential step of Verifying Required Documentation Status. By ensuring that all calibration certificates, maintenance logs, and standard operating procedures (SOPs) are present and up-to-date before the technician arrives on-site, the organization can prevent delays and identify documentation gaps before they escalate into critical audit findings. This proactive approach to resource allocation minimizes downtime and sets the stage for a high-integrity inspection.
Initiating the Process: Retrieving Equipment Lists
The foundation of a successful Good Manufacturing Practice (GMP) inspection lies in the accuracy of its starting point. The workflow begins with the critical step of Retrieving the Equipment List, a process that ensures no single piece of machinery or critical instrument is overlooked. In a highly regulated pharmaceutical environment, the scope of an inspection is defined by the inventory of assets that directly impact product quality and safety.
This initial phase involves pulling real-time data from the facility's Asset Management System or Computerized Maintenance Management System (CMMS). The retrieved list must be comprehensive, encompassing everything from high-precision analytical balances and bioreactors to simple temperature sensors and HVAC components. By systematically pulling an up-to-date inventory, the inspection team can ensure that the audit scope is current, accounting for newly commissioned equipment and identifying any retired assets that should no longer be under scrutiny. This step effectively sets the boundaries for the entire inspection cycle, ensuring that the subsequent technician assignments and checklist applications are applied to the correct, validated assets.
Task Delegation: Assigning Inspection Roles to Technicians
In a high-stakes GMP environment, the precision of an inspection is heavily dependent on the expertise of the individual performing the task. Once the initial scope is defined through the retrieval of the equipment list, the next critical step in the workflow is the formal assignment of inspection tasks to qualified technicians.
Effective delegation is not merely about distributing workload; it is about matching specific technical competencies with the complexity of the equipment being audited. By systematically assigning tasks within a centralized management system, organizations ensure that every technician is provided with clear, unambiguous instructions, specific timeframes, and the necessary digital checklists. This structured approach to task delegation eliminates ambiguity, reduces the risk of human error, and creates a transparent audit trail that is essential for maintaining regulatory compliance and ensuring that every piece of critical infrastructure undergoes a rigorous, standardized evaluation.
Phase 2: On-Site Inspection and Data Capture
Once the inspection team arrives on-site, the workflow transitions from preparation to active execution. This phase is the most critical stage of the process, as it involves the direct physical assessment of laboratory and manufacturing environments to ensure all processes align with established GMP standards.
The core of this phase begins with the Retrieval of the Equipment List, ensuring the inspectors have an accurate, real-time inventory of all assets slated for review. Once the scope is confirmed, the supervisor will Assign Inspection Tasks to Technicians, distributing specific zones or high-risk equipment to qualified personnel to ensure comprehensive coverage.
As technicians move through the facility, the focus shifts to real-time documentation. Inspectors will Log Inspection Checklist Findings directly into the digital workflow, capturing granular details regarding cleanliness, environmental controls, and operational adherence. During this process, the system will simultaneously Verify Required Documentation Status, cross-referencing physical observations with logs, SOPs, and training records to identify any gaps in the paper trail.
As discrepancies are identified, the workflow moves into automated assessment. The system will Calculate a Non-Conformance Score based on the severity and frequency of the observed deviations, providing an immediate quantitative metric of compliance health. This data-driven approach ensures that potential risks are quantified instantly rather than waiting for a post-audit review.
Real-Time Documentation: Logging Inspection Checklist Findings
In a pharmaceutical manufacturing environment, the integrity of an audit depends entirely on the accuracy and immediacy of data entry. The step of Logging Inspection Checklist Findings serves as the critical juncture where physical observations are transformed into actionable digital records.
Moving away from paper-based logs eliminates the risks of illegible handwriting, lost sheets, and retrospective data entry-all of which are significant red flags during a regulatory inspection. By utilizing a real-time digital workflow, technicians can record findings directly on-site, ensuring that every deviation, minor anomaly, or compliant parameter is captured the moment it is observed. This immediacy ensures that the data reflects the true state of the equipment and the facility, providing a single source of truth that is essential for maintaining a state of continuous GMP compliance.
Phase 3: Risk Assessment and Quantitative Analysis
Once the initial inspection findings have been recorded, the workflow transitions from data collection to a critical analytical phase. During this stage, the focus shifts from qualitative observation to quantitative assessment to ensure that every deviation is appropriately prioritized. The core of this phase involves two critical sub-processes: Calculating the Non-Conformance Score and Aggregating All Inspection Observations.
The calculation of the Non-Conformance Score is the engine of the risk assessment process. Rather than treating every discrepancy with the same level of urgency, this step applies a weighted mathematical approach to the logged findings. By evaluating the severity, frequency, and potential impact of each logged issue on product quality and patient safety, the system assigns a numerical value to the risk level. This allows quality assurance teams to distinguish between minor clerical errors and critical breaches of Good Manufacturing Practice (GMP) that could compromise sterilization or potency.
Simultaneously, the workflow moves to aggregate all inspection observations into a unified data set. This aggregation is vital for identifying systemic trends that might not be visible when looking at a single checklist in isolation. By consolidating observations across different pieces of equipment and different inspection cycles, the system provides a macro-level view of the facility's compliance health. This high-level oversight is what enables the transition from reactive troubleshooting to proactive, data-driven quality management.
Quantifying Deviations: Calculating Non-Conformance Scores
In the rigorous landscape of pharmaceutical manufacturing, a qualitative observation is often insufficient for high-level decision-making. To transform raw inspection data into actionable intelligence, the workflow incorporates a critical quantitative step: Calculating the Non-Conformance Score.
Rather than treating every discrepancy as an isolated event, this automated step applies a weighted scoring logic to the findings logged during the inspection. Each deviation is categorized based on its severity-ranging from minor housekeeping issues to critical breaches of Good Manufacturing Practices (GMP) that could compromise product sterility or patient safety.
By assigning numerical values to these varying levels of risk, the system generates a standardized metric that allows Quality Assurance (QA) teams to:
- Prioritize Resources: Instantly identify which equipment or production lines require immediate intervention versus those that only need routine monitoring.
- Identify Trends: Move beyond looking at single inspections to spotting systemic failures across entire facilities through longitudinal data analysis.
- Benchmark Performance: Establish a measurable baseline for compliance health that can be tracked through quarterly or annual audits.
This mathematical approach removes the subjectivity often found in manual auditing, ensuring that the transition from finding an error to initiating a corrective action is driven by data-driven urgency and standardized risk assessment.
Phase 4: Post-Inspection Remediation and Updates
Once the initial inspection findings are documented, the workflow shifts from observation to active remediation. This phase is critical for maintaining a state of control and ensuring that any deviations identified during the audit are addressed before they can escalate into systemic compliance failures.
The process begins with the automated Generation of Corrective Action Plan (CAPA) Tasks. Based on the documented discrepancies, the system triggers specific tasks assigned to relevant department heads, ensuring that every non-conformance is met with a structured, time-bound resolution strategy. Simultaneously, the system continues to Aggregate All Inspection Observations from across the facility, providing a holistic view of potential trends or recurring issues that might indicate a breakdown in larger quality processes.
To ensure the integrity of the equipment being audited, the workflow automatically triggers an Update to Equipment Calibration Status. This ensures that if an inspection reveals a calibration drift, the equipment's digital twin is updated immediately to prevent unauthorized use.
Crucially, this phase involves a rigorous Verification of Required Documentation Status. This step ensures that all necessary logs, certificates, and training records associated with the inspection have been reviewed and are audit-ready. By closing the loop between finding a discrepancy and verifying the documentation of its resolution, the organization ensures a closed-loop quality system that satisfies both internal standards and stringent regulatory expectations.
Maintaining Equipment Integrity: Updating Calibration Status
A critical milestone in the inspection workflow occurs immediately following the evaluation of findings: Updating Equipment Calibration Status. In a GMP-regulated environment, the integrity of your manufacturing process is directly tied to the precision of your instruments. Once the inspection checklist has been logged and the non-conformance scores calculated, the system must automatically reflect the current state of every piece of equipment involved.
This step serves as the single source of truth for your facility. If an inspection reveals that an instrument is operating outside of predefined tolerances, the calibration status must be updated to Out of Calibration or Service Required in real-time. This prevents the accidental use of compromised equipment in production, mitigating the risk of batch failures or regulatory citations. By seamlessly integrating inspection outcomes with your calibration database, you ensure that your equipment roster is always accurate, audit-ready, and fundamentally compliant with validated standards.
Closing the Loop: Generating Corrective Action Plans (CAPA)
An inspection loses its value if the findings do not lead to measurable improvements. In a GMP-regulated environment, the transition from identifying a non-conformance to implementing a solution is the most critical phase of the workflow. Once the Calculate Non-Conformance Score step identifies a deviation that exceeds acceptable thresholds, the system must automatically trigger the Generate Corrective Action Plan Task step.
This automated transition ensures that no observation is left unaddressed. Instead of relying on manual follow-ups, the workflow proactively assigns specific remediation tasks to responsible personnel, complete with predefined deadlines and required evidence of resolution. By integrating the CAPA generation directly into the inspection lifecycle, organizations can move beyond simple detection and toward a state of continuous compliance, ensuring that every gap in equipment calibration or procedural adherence is met with a documented, verifiable, and timely corrective action.
Phase 5: Reporting, Verification, and Stakeholder Communication
Once the inspection is complete and the non-conformance scores have been calculated, the workflow transitions into its final, most critical stage: formalizing the findings and ensuring accountability. This phase is designed to transform raw inspection data into actionable intelligence and verifiable compliance records.
The process begins with the generation of the Final Audit Report, which serves as the official record of the inspection. This report synthesizes all gathered data to provide a clear picture of the equipment's current state. Simultaneously, the system aggregates all inspection observations recorded throughout the process, ensuring that no detail-from minor deviations to major non-conformities-is overlooked.
To ensure the integrity of the audit, the workflow triggers a verification of required documentation status. This step confirms that all necessary certificates, logs, and calibration records are present and audit-ready, closing any gaps in the evidentiary trail.
The final step in the loop is the generation of Corrective Action Plan (CAPA) tasks. By automatically converting identified non-conformances into assigned tasks, the workflow ensures that finding a problem is immediately followed by fixing a problem. To close the loop, the system automatically notifies all relevant stakeholders of the inspection completion. This automated communication ensures that Quality Assurance managers, maintenance teams, and department heads are instantly informed of the results, fostering a culture of transparency and rapid response in maintaining GMP standards.
Conclusion: Ensuring Audit Readiness through Systematic Management
Mastering the Pharmaceutical GMP Inspection Workflow: Validated Compliance and Audit Management is not merely about passing a single inspection; it is about embedding a culture of continuous compliance within your facility. By transitioning from reactive troubleshooting to a proactive, systematic approach-where every step from retrieving equipment lists to notifying stakeholders is standardized-you eliminate the chaos often associated with regulatory audits.
A structured workflow ensures that non-conformances are not just recorded, but are quantified, addressed through automated corrective action plans, and integrated into a comprehensive data aggregate. This level of visibility allows quality managers to identify recurring trends before they escalate into critical findings. Ultimately, by digitizing and automating these inspection touchpoints, you transform your quality management system into a robust, audit-ready powerhouse that guarantees both equipment reliability and unwavering regulatory compliance.
Resources & Links
- U.S. Food and Drug Administration (FDA): Official regulatory guidelines and resources regarding Good Manufacturing Practices (GMP) and inspection standards for pharmaceutical products.
- European Medicines Agency (EMA): Regulatory information regarding EU GMP standards, compliance requirements, and inspection protocols for the European market.
- International Society for Pharmaceutical Engineering (ISPE): Professional resources and industry standards for pharmaceutical manufacturing, focusing on technical compliance and workflow excellence.
- World Health Organization (WHO): Global standards for Good Manufacturing Practices and guidelines for quality control and inspection processes in pharmaceutical manufacturing.
- Pharmaceutical Quality Systems (PQS) Framework: Foundational principles regarding Quality Management Systems (QMS) and the integration of CAPA and risk management in audits.
- International Organization for Standardization (ISO): Information on ISO standards related to quality management (ISO 9001) and laboratory testing (ISO/IEC 17025) applicable to inspection workflows.
Found this Article helpful?
Audit/Inspection Management Solution Demo
Ensure compliance & improve performance! ChecklistGuro streamlines audit/inspection creation, execution, and reporting. Reduce risk, enhance quality, & maintain consistency. Manage it all with our Work OS.
Related Articles
Restaurant Kitchen Safety Inspection Checklist Template
The Ultimate Brewery Equipment Sanitation & Safety Checklist Template
Wind Turbine Inspection Checklist Template: Your Comprehensive Guide
The Ultimate Apartment Building Maintenance Inspection Checklist Template
Your Essential Guide to a Manufacturing Quality Control Inspection Checklist
The Ultimate Solar Panel Inspection Checklist Template
Your Ultimate HVAC Inspection Checklist Template
The Ultimate Data Center Inspection Checklist Template
We can do it Together
Need help with
Checklists?
Have a question? We're here to help. Please submit your inquiry, and we'll respond promptly.